-
 蘇州新藥eCTD
蘇州新藥eCTD歐洲YAO品管理局:集中審評程序由歐洲YAO品管理局(EuropeanMedicinesAgency,EMA)負責協調。人用YAO品委YUAN會:人用YAO品委YUAN會(CommitteeforMedicinalProductsforHumanU...
2025-12-20 -
 無錫賦悅科技eCTD找哪家
無錫賦悅科技eCTD找哪家GDUFAIII框架與費用分類2022年更的GDUFAIII將費用分為ANDA申請費、DMF認證費、項目費及設施費四類,實施周期至2027年。2025財年ANDA費用長至約22萬美元,較2024年增幅達,反映審評成本上升。ANDA申請費規則費用需在...
2025-12-20 -
 浙江仿制藥eCTD哪個品牌好
浙江仿制藥eCTD哪個品牌好Z國將進一步與guo際接軌,推進eCTD等標準應用,提高yao品注冊效率和質量。AI技術可能在yao品注冊領域廣泛應用,如輔助審評人員工作。未來yao品注冊資料將更注重結構化數據,有助于監管機構gao效獲取和利用數據。eCTD等數字化工具將推動ya...
2025-12-20 -
 蕪湖新藥eCTD服務放心可靠
蕪湖新藥eCTD服務放心可靠eCTD文件制作需遵循嚴格的法規要求和標準化流程,以下是關鍵要點整理:eCTD采用模塊化結構,包含模塊1(行政信息)至模塊5(臨床報告),需按ICH和監機構要求構建目錄樹。顆粒度選擇:文件提交層級需在***申報時確定并沿用,例如原料和制劑的章節(如、)需按比較...
2025-12-20 -
 寧波電子申報eCTD服務電話
寧波電子申報eCTD服務電話美國于2003年成為全球早采用eCTD(電子通用技術文檔)的國家之一,初由CDER和CBER作為電子提交平臺試點。2008年起,eCTD正式成為藥申請(NDA)和生物制品許可申請(BLA)的標準格式,并在2012年通過《藥申報者付費法案》(PDUFA)進一步強...
2025-12-19 -
 工業園區原料藥eCTD系統
工業園區原料藥eCTD系統美國于2003年成為全球早采用eCTD(電子通用技術文檔)的國家之一,初由CDER和CBER作為電子提交平臺試點。2008年起,eCTD正式成為藥申請(NDA)和生物制品許可申請(BLA)的標準格式,并在2012年通過《藥申報者付費法案》(PDUFA)進一步強...
2025-12-19 -
 楊浦區化學藥品eCTD常用解決方案
楊浦區化學藥品eCTD常用解決方案生命周期管理與變更遞交eCTD支持全生命周期管理,申請人需通過序列更(Sequence)反映yao品變更信息。例如CEP的更需提交“變更說明表”,對比已批準和擬修改內容,并附修訂版技術文檔。重大變更(如生產工藝調整)可能觸發GMP現場檢查,EDQM...
2025-12-19 -
江蘇中國eCTD遞交
eCTD:FDA于2023年啟動eCTD,2024年9月正式接收申請,計劃2029年完成全過渡。RPS標準替代XML,支持雙向通信和跨申請文件復用,例如同一StudyID可在IND和NDA享。模塊1的校驗碼從MD5升級為SHA-256,主干文件由,...
2025-12-19 -
 蘇州ANDAeCTD哪個品牌好
蘇州ANDAeCTD哪個品牌好緊急申報與特殊通道:FDA設置緊急申報通道(如Pre-EUA和EUA),允許在公共衛生事件中快su提交資料。此類申請需在模塊,并通過ESG加急處理。eCTD版本兼容性與過渡策略:eCTD,允許。企業需在2024年前完成系統升級,確保XML到HL7R...
2025-12-19 -
 吳江區賦悅科技eCTD使用
吳江區賦悅科技eCTD使用仿制yao作為提高yao物可及性與可負擔性的一類yao物,2012年以前,注冊審評是不收取任何費用的,但當時仿制yao申請積壓嚴重,從申報到獲批需要3~5年的時間。美國國會于2012年頒布了仿制yao使用者費用修正案(GenericDrugUser...
2025-12-19 -
 浦東新區中國eCTD使用
浦東新區中國eCTD使用eCTD的實施為監管機構和企業帶來了多重機遇。電子化申報資料能夠極大地加速審評效率,減少人為判斷錯誤和數據混淆的情況,從而提高審評的準確性和速度。同時,eCTD帶來的數據標準化機遇使得全球監管機構的資料內容和電子格式得以統一,有助于在不同監管機構之間進行數據傳...
2025-12-19 -
 上海化學藥品eCTD品牌
上海化學藥品eCTD品牌內容與格式檢查Word預處理:需檢查拼寫、縮略語、單位格式(如),設置多級列表自動編號(如),統一字體(宋體/TimesNewRoman)和段落格式。重復內容處理:相同劑型不同規格可共用模塊3,但需區分包裝系統(如、)。外文資料:中文在前、原文在后,參考文獻需...
2025-12-19 -
浦東新區仿制藥eCTD服務電話
Z國將進一步與guo際接軌,推進eCTD等標準應用,提高yao品注冊效率和質量。AI技術可能在yao品注冊領域廣泛應用,如輔助審評人員工作。未來yao品注冊資料將更注重結構化數據,有助于監管機構gao效獲取和利用數據。eCTD等數字化工具將推動ya...
2025-12-19 -
 靜安區仿制藥eCTD哪個品牌好
靜安區仿制藥eCTD哪個品牌好eCTD即是電子化的CTD注冊申報方式,相對于傳統的紙質遞交,eCTD電子遞交更便捷、更安全。對于申請者來說,一個產品如在多個市場遞交,M2-M5的資料可以共享,極大降低了成本并提高了效率;對審評者來說,eCTD資料的審閱、管理、傳輸以及歸檔十分便...
2025-12-19 -
蘇州化學藥品eCTD醫療科技
eCTD即是電子化的CTD注冊申報方式,相對于傳統的紙質遞交,eCTD電子遞交更便捷、更安全。對于申請者來說,一個產品如在多個市場遞交,M2-M5的資料可以共享,極大降低了成本并提高了效率;對審評者來說,eCTD資料的審閱、管理、傳輸以及歸檔十分便...
2025-12-19 -
無錫仿制藥eCTD服務商
eCTD的實施為監管機構和企業帶來了多重機遇。電子化申報資料能夠極大地加速審評效率,減少人為判斷錯誤和數據混淆的情況,從而提高審評的準確性和速度。同時,eCTD帶來的數據標準化機遇使得全球監管機構的資料內容和電子格式得以統一,有助于在不同監管機構之間進行數...
2025-12-19 -
工業園區INDeCTD報價
緊急申報與特殊通道:FDA設置緊急申報通道(如Pre-EUA和EUA),允許在公共衛生事件中快su提交資料。此類申請需在模塊,并通過ESG加急處理。eCTD版本兼容性與過渡策略:eCTD,允許。企業需在2024年前完成系統升級,確保XML到HL7R...
2025-12-19 -
 遼寧仿制藥eCTD
遼寧仿制藥eCTD區域化差異與多國協作挑戰 歐盟eCTD需兼容成員國特定要求,例如模塊一的行政信息需符合各國語言和法規差異。互認程序(MRP)中,參考成員國(RMS)的評估報告需被其他成員國認可,若出現分歧需由CMDh協調或提交EMA仲裁。這種多層級審評機制要求申請人在文件準備...
2025-12-19 -
合肥原料藥eCTD哪家好
eCTD:FDA于2023年啟動eCTD,2024年9月正式接收申請,計劃2029年完成全過渡。RPS標準替代XML,支持雙向通信和跨申請文件復用,例如同一StudyID可在IND和NDA享。模塊1的校驗碼從MD5升級為SHA-256,主干文件由,...
2025-12-19 -
 杭州國際注冊eCTD歡迎選購
杭州國際注冊eCTD歡迎選購從紙質到電子的歷史過渡 2017年前,美國允許紙質與eCTD并行提交,但此后逐步淘汰紙質通道,保留緊急情況下的例外審批。2020年電子化后,所有IND、NDA、ANDA和DMF強制采用eCTD格式。 系統平臺升級 FDA通過“yao品業務應用系統”和“yao品...
2025-12-19 -
河北國際注冊eCTD
電子遞交的合規性與FENG險管理歐盟要求申請人確保電子資料與紙質版本完全一致,若未在規定時間提交紙質文件可能導致注冊終止。驗證過程中,“錯誤”級別問題(如文件命名不規范、XML邏輯錯誤)必須修正,而“警告”和“提示信息”則建議優化以提升審評體驗。EDQM...
2025-12-19 -
 閔行區NDAeCTD便宜
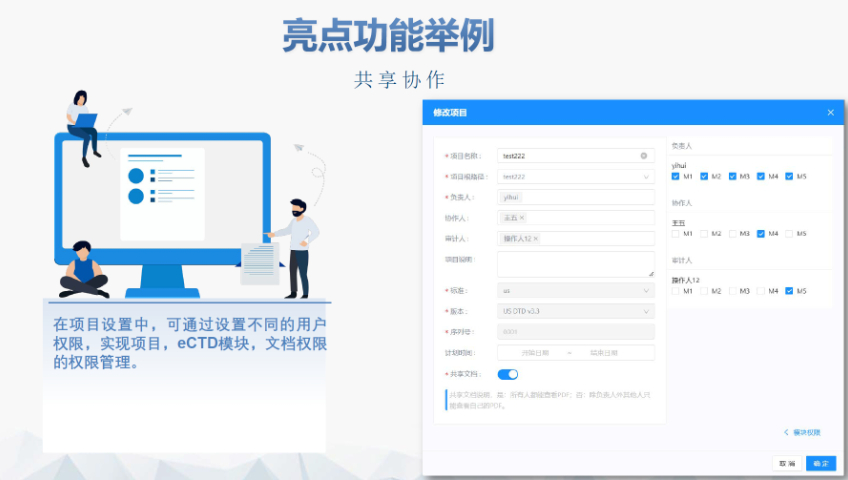
閔行區NDAeCTD便宜法規文檔管理系統協同共享RDMS可以讓跨區域、跨部門協同真正成為1+1>2的工作。讓頻繁的文檔共享傳輸,版本管理,生命周期審批都變得輕松簡單安全合規通過詳細的審計追蹤、電子簽名、權限管理、網關控zhi、頁面控zhi等技術手段,構建安全合規的文檔管理系統,...
2025-12-19 -
安徽CDE eCTD推薦
美國于2003年成為全球早采用eCTD(電子通用技術文檔)的國家之一,初由CDER和CBER作為電子提交平臺試點。2008年起,eCTD正式成為藥申請(NDA)和生物制品許可申請(BLA)的標準格式,并在2012年通過《藥申報者付費法案》(PDUFA)進一步強...
2025-12-18 -
 浦東新區電子申報eCTD服務價格
浦東新區電子申報eCTD服務價格2015年發布《關于yao品醫療器械審評審批制度的意見》,提出yao監五大目標,將eCTD納入guo家yao監數字化戰略。2017年,中guo加入ICH(guo際人用yao品注冊技術協調會),成為全球第八個監管機構成員,加速與guo際標準接軌。2018年...
2025-12-18 -
靜安區INDeCTD名稱
歐洲YAO品管理局:集中審評程序由歐洲YAO品管理局(EuropeanMedicinesAgency,EMA)負責協調。人用YAO品委YUAN會:人用YAO品委YUAN會(CommitteeforMedicinalProductsforHumanU...
2025-12-18 -
 蕪湖生物制品eCTD名稱
蕪湖生物制品eCTD名稱審評效率與時間線優化eCTD的標準化縮短了審評周期:集中程序平均審評時間從18個月降至12個月,互認程序可在90天內完成成員國意見協調。自動化驗證工具減少了格式錯誤導致的退審率,但復雜yao學數據的科學審評仍需較長時間。申請人可通過預提交會議(Pr...
2025-12-18 -
寧波NDAeCTD格式
ANDA一般不需要提供臨床前(動物)和臨床(人體)數據來證明其安全性和youxiao性(即免毒理和臨床),作為替代,申請人必須合理證明其產品與原研YAO相比是shengwu等效的。按照《聯邦食品、YAO品和化妝品法》第505(j)章要求,擬向FDA遞交申請AN...
2025-12-18 -
無錫NDAeCTD服務商
ANDA一般不需要提供臨床前(動物)和臨床(人體)數據來證明其安全性和youxiao性(即免毒理和臨床),作為替代,申請人必須合理證明其產品與原研YAO相比是shengwu等效的。按照《聯邦食品、YAO品和化妝品法》第505(j)章要求,擬向FDA遞交申請AN...
2025-12-18 -
合肥NDAeCTD發布軟件
電子簽章與安全性FDA要求所有PDF文件需經數字簽名,并通過MD5校驗確保傳輸完整性。簽章需符合21CFRPart11的電子記錄規范,部分情況下允許臨時放寬(如期間的遠程簽署)。多模塊協同驗證模塊1(行政文件)的區域性元數據(如申請類型、聯系人信息)需與模塊2...
2025-12-18 -
楊浦區新藥eCTD歡迎選購
澳大利亞的yao品電子通用技術文檔(eCTD)注冊申報體系是澳大利亞yao品商品管理局(TGA)推動yao品審評現代化的重要舉措。eCTD作為guo際通行的電子化注冊申報標準,通過結構化數據格式(如XML)整合了yao品質量、安全性和you效性的技...
2025-12-18